TCGA: Data Processing, Checking, and Exploration
Last updated: 2021-01-14
Checks: 7 0
Knit directory: esoph-micro-cancer-workflow/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200916) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version e6b4c7f. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/data_processing_tcga.Rmd) and HTML (docs/data_processing_tcga.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 1d24c1f | noah-padgett | 2020-12-17 | make waterfall-plots |
| html | 1d24c1f | noah-padgett | 2020-12-17 | make waterfall-plots |
| Rmd | cf91029 | noah-padgett | 2020-12-02 | updated analyses |
| html | cf91029 | noah-padgett | 2020-12-02 | updated analyses |
| Rmd | 9937a7e | noah-padgett | 2020-11-05 | new abundance data results |
| html | 9937a7e | noah-padgett | 2020-11-05 | new abundance data results |
| Rmd | b5c01ed | noah-padgett | 2020-10-22 | fixed TCGA figure |
| html | b5c01ed | noah-padgett | 2020-10-22 | fixed TCGA figure |
| Rmd | e41080d | noah-padgett | 2020-10-22 | updated cleaning and ids data |
| html | e41080d | noah-padgett | 2020-10-22 | updated cleaning and ids data |
| Rmd | 5b186b4 | noah-padgett | 2020-10-08 | fixed image showing |
| html | 5b186b4 | noah-padgett | 2020-10-08 | fixed image showing |
| html | 67ac872 | noah-padgett | 2020-09-24 | Build site. |
| Rmd | 498a050 | noah-padgett | 2020-09-24 | updated data processing |
| Rmd | ec3d151 | noah-padgett | 2020-09-24 | updated processing files |
For the TCGA data, data need to be processed twice. First, for the RNAseq microbiome data. Next, for the WGS microbiome data. When you try to do both at the same time, then there is a mismatch among cases with respect to the number of samples that were generated for each case.
This page contains the investigation of the raw data (OTUs) to identify if outliers are present or whether other issues emerge that may influence our results in unexpected ways. This file goes through the following checks:
- Removal of Phylum NA features
- Computation of total and average prevalence in each Phylum
- Removal Phyla with 1% or less of all samples
- Computation of total read count for each Phyla
- Plotting taxa prevalence vs total counts - identify a natural threshold if clear, if not use 5%
- Merging taxa to genus rank/level
- Abundance Value Transformations
- Plotting of abundance values by “SampleType_Level2: Tumor, Normal” before transformation and after
RNAseq Data: Taxonomic Filtering
0. Sample Reads, Totals, and Rarifying
sampleReads <- phyloseq::sample_sums(phylo.data.tcga.RNAseq)
sampleReads TCGA.2H.A9GF.Tumor.RNAseq.579 TCGA.2H.A9GG.Tumor.RNAseq.755
2196746 0
TCGA.2H.A9GH.Tumor.RNAseq.2a3 TCGA.2H.A9GI.Tumor.RNAseq.eed
0 0
TCGA.2H.A9GJ.Tumor.RNAseq.a43 TCGA.2H.A9GK.Tumor.RNAseq.942
2258184 0
TCGA.2H.A9GL.Tumor.RNAseq.82d TCGA.2H.A9GM.Tumor.RNAseq.886
0 0
TCGA.2H.A9GN.Tumor.RNAseq.c45 TCGA.2H.A9GO.Tumor.RNAseq.033
0 0
TCGA.2H.A9GQ.Tumor.RNAseq.d23 TCGA.2H.A9GR.Tumor.RNAseq.397
0 0
TCGA.IC.A6RE.Normal.RNAseq.eb2 TCGA.IC.A6RE.Tumor.RNAseq.6c9
0 0
TCGA.IC.A6RF.Normal.RNAseq.639 TCGA.IC.A6RF.Tumor.RNAseq.5d9
0 0
TCGA.IG.A3I8.Tumor.RNAseq.f83 TCGA.IG.A3QL.Tumor.RNAseq.85d
208954 91069
TCGA.IG.A3YA.Tumor.RNAseq.66a TCGA.IG.A3YB.Tumor.RNAseq.8fe
57093 0
TCGA.IG.A3YC.Tumor.RNAseq.276 TCGA.IG.A4P3.Tumor.RNAseq.50d
581915 309817
TCGA.IG.A4QS.Tumor.RNAseq.542 TCGA.IG.A50L.Tumor.RNAseq.93f
964946 486713
TCGA.IG.A51D.Tumor.RNAseq.feb TCGA.IG.A5B8.Tumor.RNAseq.3b2
0 677270
TCGA.IG.A5S3.Tumor.RNAseq.200 TCGA.IG.A625.Tumor.RNAseq.096
195220 143832
TCGA.IG.A6QS.Tumor.RNAseq.817 TCGA.IG.A7DP.Tumor.RNAseq.ba3
0 0
TCGA.IG.A8O2.Tumor.RNAseq.982 TCGA.IG.A97H.Tumor.RNAseq.1d7
0 0
TCGA.IG.A97I.Tumor.RNAseq.432 TCGA.JY.A6FA.Tumor.RNAseq.f91
0 0
TCGA.JY.A6FB.Tumor.RNAseq.6b6 TCGA.JY.A6FD.Tumor.RNAseq.339
0 0
TCGA.JY.A6FE.Tumor.RNAseq.3bf TCGA.JY.A6FG.Tumor.RNAseq.cf4
0 0
TCGA.JY.A6FH.Tumor.RNAseq.ede TCGA.JY.A938.Tumor.RNAseq.385
0 0
TCGA.JY.A939.Tumor.RNAseq.9a2 TCGA.JY.A93C.Tumor.RNAseq.3ba
0 1912961
TCGA.JY.A93D.Tumor.RNAseq.2c3 TCGA.JY.A93E.Tumor.RNAseq.2a0
0 0
TCGA.JY.A93F.Tumor.RNAseq.f8f TCGA.KH.A6WC.Tumor.RNAseq.743
0 0
TCGA.L5.A43C.Normal.RNAseq.ffd TCGA.L5.A43C.Tumor.RNAseq.5fe
434352 751188
TCGA.L5.A43E.Tumor.RNAseq.6bb TCGA.L5.A43J.Tumor.RNAseq.64f
148331 31941
TCGA.L5.A4OE.Tumor.RNAseq.0e5 TCGA.L5.A4OF.Normal.RNAseq.4cb
242287 281031
TCGA.L5.A4OG.Normal.RNAseq.76d TCGA.L5.A4OG.Tumor.RNAseq.ef4
449297 212212
TCGA.L5.A4OH.Tumor.RNAseq.0ce TCGA.L5.A4OI.Tumor.RNAseq.1fe
242003 0
TCGA.L5.A4OJ.Normal.RNAseq.d64 TCGA.L5.A4OJ.Tumor.RNAseq.17c
413474 236190
TCGA.L5.A4OM.Tumor.RNAseq.9d2 TCGA.L5.A4ON.Tumor.RNAseq.2e8
161300 94813
TCGA.L5.A4OO.Normal.RNAseq.646 TCGA.L5.A4OO.Tumor.RNAseq.1f1
462884 364963
TCGA.L5.A4OP.Tumor.RNAseq.6be TCGA.L5.A4OQ.Normal.RNAseq.c24
293979 213727
TCGA.L5.A4OR.Normal.RNAseq.22f TCGA.L5.A4OS.Tumor.RNAseq.85f
374051 233892
TCGA.L5.A4OT.Tumor.RNAseq.71d TCGA.L5.A4OU.Tumor.RNAseq.df3
507148 433111
TCGA.L5.A4OW.Tumor.RNAseq.e8e TCGA.L5.A4OX.Tumor.RNAseq.c54
317861 238826
TCGA.L5.A88S.Tumor.RNAseq.906 TCGA.L5.A88T.Tumor.RNAseq.e81
0 0
TCGA.L5.A88V.Tumor.RNAseq.ecb TCGA.L5.A88W.Tumor.RNAseq.2b3
0 0
TCGA.L5.A88Y.Tumor.RNAseq.9f3 TCGA.L5.A88Z.Tumor.RNAseq.6c5
0 0
TCGA.L5.A891.Tumor.RNAseq.b37 TCGA.L5.A893.Tumor.RNAseq.4df
0 0
TCGA.L5.A8NE.Tumor.RNAseq.0e5 TCGA.L5.A8NF.Tumor.RNAseq.42a
0 0
TCGA.L5.A8NG.Tumor.RNAseq.106 TCGA.L5.A8NH.Tumor.RNAseq.e87
0 0
TCGA.L5.A8NI.Tumor.RNAseq.5da TCGA.L5.A8NJ.Tumor.RNAseq.fc1
0 0
TCGA.L5.A8NK.Tumor.RNAseq.2e1 TCGA.L5.A8NL.Tumor.RNAseq.702
0 0
TCGA.L5.A8NM.Tumor.RNAseq.8c6 TCGA.L5.A8NN.Tumor.RNAseq.b33
0 0
TCGA.L5.A8NQ.Tumor.RNAseq.e97 TCGA.L5.A8NR.Tumor.RNAseq.05b
0 0
TCGA.L5.A8NS.Tumor.RNAseq.69a TCGA.L5.A8NT.Tumor.RNAseq.8c4
2377000 0
TCGA.L5.A8NU.Tumor.RNAseq.411 TCGA.L5.A8NV.Tumor.RNAseq.9c7
0 0
TCGA.L5.A8NW.Tumor.RNAseq.023 TCGA.L7.A56G.Tumor.RNAseq.70a
0 328304
TCGA.L7.A6VZ.Tumor.RNAseq.f69 TCGA.LN.A49M.Tumor.RNAseq.d16
0 269249
TCGA.LN.A49O.Tumor.RNAseq.d4f TCGA.LN.A49P.Tumor.RNAseq.346
18157 60432
TCGA.LN.A49S.Tumor.RNAseq.0a9 TCGA.LN.A49U.Tumor.RNAseq.450
55311 183716
TCGA.LN.A49W.Tumor.RNAseq.dfd TCGA.LN.A49X.Tumor.RNAseq.36d
293817 266304
TCGA.LN.A49Y.Tumor.RNAseq.32b TCGA.LN.A4A1.Tumor.RNAseq.ffd
589419 198953
TCGA.LN.A4A3.Tumor.RNAseq.7ab TCGA.LN.A4A4.Tumor.RNAseq.fc5
330219 345745
TCGA.LN.A4A5.Tumor.RNAseq.11e TCGA.LN.A4A8.Tumor.RNAseq.e2a
167082 169882
TCGA.LN.A4A9.Tumor.RNAseq.cc6 TCGA.LN.A4MQ.Tumor.RNAseq.5b4
498383 210714
TCGA.LN.A5U5.Tumor.RNAseq.ef4 TCGA.LN.A5U6.Tumor.RNAseq.c00
208664 205554
TCGA.LN.A5U7.Tumor.RNAseq.88b TCGA.LN.A7HV.Tumor.RNAseq.504
111690 0
TCGA.LN.A7HW.Tumor.RNAseq.1ad TCGA.LN.A7HX.Tumor.RNAseq.99e
0 0
TCGA.LN.A7HY.Tumor.RNAseq.f7b TCGA.LN.A7HZ.Tumor.RNAseq.740
0 0
TCGA.LN.A8HZ.Tumor.RNAseq.628 TCGA.LN.A8I0.Tumor.RNAseq.b5f
0 0
TCGA.LN.A8I1.Tumor.RNAseq.838 TCGA.LN.A9FO.Tumor.RNAseq.ae5
0 0
TCGA.LN.A9FP.Tumor.RNAseq.350 TCGA.LN.A9FQ.Tumor.RNAseq.082
0 0
TCGA.LN.A9FR.Tumor.RNAseq.304 TCGA.M9.A5M8.Tumor.RNAseq.4ef
0 233106
TCGA.Q9.A6FW.Tumor.RNAseq.d88 TCGA.R6.A6DN.Tumor.RNAseq.7ed
472304 197164
TCGA.R6.A6DQ.Tumor.RNAseq.a41 TCGA.R6.A6KZ.Tumor.RNAseq.22d
302461 188388
TCGA.R6.A6L4.Tumor.RNAseq.19e TCGA.R6.A6XG.Tumor.RNAseq.fde
242143 0
TCGA.R6.A6XQ.Tumor.RNAseq.ce6 TCGA.R6.A6Y0.Tumor.RNAseq.815
0 0
TCGA.R6.A8W5.Tumor.RNAseq.300 TCGA.R6.A8W8.Tumor.RNAseq.fb2
0 2269286
TCGA.R6.A8WC.Tumor.RNAseq.64b TCGA.R6.A8WG.Tumor.RNAseq.cb5
2118504 0
TCGA.RE.A7BO.Tumor.RNAseq.e67 TCGA.S8.A6BV.Tumor.RNAseq.86b
0 215598
TCGA.S8.A6BW.Tumor.RNAseq.802 TCGA.V5.A7RB.Tumor.RNAseq.2c7
136204 0
TCGA.V5.A7RC.Tumor.RNAseq.15e TCGA.V5.A7RC.Tumor.RNAseq.5f5
0 0
TCGA.V5.A7RE.Normal.RNAseq.848 TCGA.V5.A7RE.Tumor.RNAseq.7c1
0 0
TCGA.V5.AASV.Tumor.RNAseq.2bb TCGA.V5.AASW.Tumor.RNAseq.a2a
0 0
TCGA.V5.AASX.Normal.RNAseq.e7e TCGA.V5.AASX.Tumor.RNAseq.0c4
0 0
TCGA.VR.A8EO.Tumor.RNAseq.e76 TCGA.VR.A8EP.Tumor.RNAseq.afe
0 0
TCGA.VR.A8EQ.Tumor.RNAseq.350 TCGA.VR.A8ER.Tumor.RNAseq.a53
0 0
TCGA.VR.A8ET.Tumor.RNAseq.d5b TCGA.VR.A8EU.Tumor.RNAseq.421
0 0
TCGA.VR.A8EW.Tumor.RNAseq.a6b TCGA.VR.A8EX.Tumor.RNAseq.547
0 0
TCGA.VR.A8EY.Tumor.RNAseq.48e TCGA.VR.A8EZ.Tumor.RNAseq.48d
0 0
TCGA.VR.A8Q7.Tumor.RNAseq.053 TCGA.VR.AA4D.Tumor.RNAseq.325
0 0
TCGA.VR.AA4G.Tumor.RNAseq.52c TCGA.VR.AA7I.Tumor.RNAseq.1a6
0 0
TCGA.XP.A8T6.Tumor.RNAseq.5ca TCGA.XP.A8T8.Tumor.RNAseq.2d9
0 0
TCGA.Z6.A8JD.Tumor.RNAseq.d53 TCGA.Z6.A8JE.Tumor.RNAseq.e1a
0 0
TCGA.Z6.A9VB.Tumor.RNAseq.f2b TCGA.Z6.AAPN.Tumor.RNAseq.64f
0 0
TCGA.ZR.A9CJ.Tumor.RNAseq.d6f
0 # Total quality Reads
sum(sampleReads)[1] 30487334# Average reads
mean(sampleReads)[1] 176227.4# max sequencing depth
max(sampleReads)[1] 2377000# min sequencing depth
min(sampleReads)[1] 0# rarified to an even depth of
#phylo.data.tcga <- phyloseq::rarefy_even_depth(phylo.data.tcga.RNAseq, replace = T, rngseed = 20200923)
phylo.data.tcga <- phylo.data.tcga.RNAseq
# even depth of:
phyloseq::sample_sums(phylo.data.tcga) TCGA.2H.A9GF.Tumor.RNAseq.579 TCGA.2H.A9GG.Tumor.RNAseq.755
2196746 0
TCGA.2H.A9GH.Tumor.RNAseq.2a3 TCGA.2H.A9GI.Tumor.RNAseq.eed
0 0
TCGA.2H.A9GJ.Tumor.RNAseq.a43 TCGA.2H.A9GK.Tumor.RNAseq.942
2258184 0
TCGA.2H.A9GL.Tumor.RNAseq.82d TCGA.2H.A9GM.Tumor.RNAseq.886
0 0
TCGA.2H.A9GN.Tumor.RNAseq.c45 TCGA.2H.A9GO.Tumor.RNAseq.033
0 0
TCGA.2H.A9GQ.Tumor.RNAseq.d23 TCGA.2H.A9GR.Tumor.RNAseq.397
0 0
TCGA.IC.A6RE.Normal.RNAseq.eb2 TCGA.IC.A6RE.Tumor.RNAseq.6c9
0 0
TCGA.IC.A6RF.Normal.RNAseq.639 TCGA.IC.A6RF.Tumor.RNAseq.5d9
0 0
TCGA.IG.A3I8.Tumor.RNAseq.f83 TCGA.IG.A3QL.Tumor.RNAseq.85d
208954 91069
TCGA.IG.A3YA.Tumor.RNAseq.66a TCGA.IG.A3YB.Tumor.RNAseq.8fe
57093 0
TCGA.IG.A3YC.Tumor.RNAseq.276 TCGA.IG.A4P3.Tumor.RNAseq.50d
581915 309817
TCGA.IG.A4QS.Tumor.RNAseq.542 TCGA.IG.A50L.Tumor.RNAseq.93f
964946 486713
TCGA.IG.A51D.Tumor.RNAseq.feb TCGA.IG.A5B8.Tumor.RNAseq.3b2
0 677270
TCGA.IG.A5S3.Tumor.RNAseq.200 TCGA.IG.A625.Tumor.RNAseq.096
195220 143832
TCGA.IG.A6QS.Tumor.RNAseq.817 TCGA.IG.A7DP.Tumor.RNAseq.ba3
0 0
TCGA.IG.A8O2.Tumor.RNAseq.982 TCGA.IG.A97H.Tumor.RNAseq.1d7
0 0
TCGA.IG.A97I.Tumor.RNAseq.432 TCGA.JY.A6FA.Tumor.RNAseq.f91
0 0
TCGA.JY.A6FB.Tumor.RNAseq.6b6 TCGA.JY.A6FD.Tumor.RNAseq.339
0 0
TCGA.JY.A6FE.Tumor.RNAseq.3bf TCGA.JY.A6FG.Tumor.RNAseq.cf4
0 0
TCGA.JY.A6FH.Tumor.RNAseq.ede TCGA.JY.A938.Tumor.RNAseq.385
0 0
TCGA.JY.A939.Tumor.RNAseq.9a2 TCGA.JY.A93C.Tumor.RNAseq.3ba
0 1912961
TCGA.JY.A93D.Tumor.RNAseq.2c3 TCGA.JY.A93E.Tumor.RNAseq.2a0
0 0
TCGA.JY.A93F.Tumor.RNAseq.f8f TCGA.KH.A6WC.Tumor.RNAseq.743
0 0
TCGA.L5.A43C.Normal.RNAseq.ffd TCGA.L5.A43C.Tumor.RNAseq.5fe
434352 751188
TCGA.L5.A43E.Tumor.RNAseq.6bb TCGA.L5.A43J.Tumor.RNAseq.64f
148331 31941
TCGA.L5.A4OE.Tumor.RNAseq.0e5 TCGA.L5.A4OF.Normal.RNAseq.4cb
242287 281031
TCGA.L5.A4OG.Normal.RNAseq.76d TCGA.L5.A4OG.Tumor.RNAseq.ef4
449297 212212
TCGA.L5.A4OH.Tumor.RNAseq.0ce TCGA.L5.A4OI.Tumor.RNAseq.1fe
242003 0
TCGA.L5.A4OJ.Normal.RNAseq.d64 TCGA.L5.A4OJ.Tumor.RNAseq.17c
413474 236190
TCGA.L5.A4OM.Tumor.RNAseq.9d2 TCGA.L5.A4ON.Tumor.RNAseq.2e8
161300 94813
TCGA.L5.A4OO.Normal.RNAseq.646 TCGA.L5.A4OO.Tumor.RNAseq.1f1
462884 364963
TCGA.L5.A4OP.Tumor.RNAseq.6be TCGA.L5.A4OQ.Normal.RNAseq.c24
293979 213727
TCGA.L5.A4OR.Normal.RNAseq.22f TCGA.L5.A4OS.Tumor.RNAseq.85f
374051 233892
TCGA.L5.A4OT.Tumor.RNAseq.71d TCGA.L5.A4OU.Tumor.RNAseq.df3
507148 433111
TCGA.L5.A4OW.Tumor.RNAseq.e8e TCGA.L5.A4OX.Tumor.RNAseq.c54
317861 238826
TCGA.L5.A88S.Tumor.RNAseq.906 TCGA.L5.A88T.Tumor.RNAseq.e81
0 0
TCGA.L5.A88V.Tumor.RNAseq.ecb TCGA.L5.A88W.Tumor.RNAseq.2b3
0 0
TCGA.L5.A88Y.Tumor.RNAseq.9f3 TCGA.L5.A88Z.Tumor.RNAseq.6c5
0 0
TCGA.L5.A891.Tumor.RNAseq.b37 TCGA.L5.A893.Tumor.RNAseq.4df
0 0
TCGA.L5.A8NE.Tumor.RNAseq.0e5 TCGA.L5.A8NF.Tumor.RNAseq.42a
0 0
TCGA.L5.A8NG.Tumor.RNAseq.106 TCGA.L5.A8NH.Tumor.RNAseq.e87
0 0
TCGA.L5.A8NI.Tumor.RNAseq.5da TCGA.L5.A8NJ.Tumor.RNAseq.fc1
0 0
TCGA.L5.A8NK.Tumor.RNAseq.2e1 TCGA.L5.A8NL.Tumor.RNAseq.702
0 0
TCGA.L5.A8NM.Tumor.RNAseq.8c6 TCGA.L5.A8NN.Tumor.RNAseq.b33
0 0
TCGA.L5.A8NQ.Tumor.RNAseq.e97 TCGA.L5.A8NR.Tumor.RNAseq.05b
0 0
TCGA.L5.A8NS.Tumor.RNAseq.69a TCGA.L5.A8NT.Tumor.RNAseq.8c4
2377000 0
TCGA.L5.A8NU.Tumor.RNAseq.411 TCGA.L5.A8NV.Tumor.RNAseq.9c7
0 0
TCGA.L5.A8NW.Tumor.RNAseq.023 TCGA.L7.A56G.Tumor.RNAseq.70a
0 328304
TCGA.L7.A6VZ.Tumor.RNAseq.f69 TCGA.LN.A49M.Tumor.RNAseq.d16
0 269249
TCGA.LN.A49O.Tumor.RNAseq.d4f TCGA.LN.A49P.Tumor.RNAseq.346
18157 60432
TCGA.LN.A49S.Tumor.RNAseq.0a9 TCGA.LN.A49U.Tumor.RNAseq.450
55311 183716
TCGA.LN.A49W.Tumor.RNAseq.dfd TCGA.LN.A49X.Tumor.RNAseq.36d
293817 266304
TCGA.LN.A49Y.Tumor.RNAseq.32b TCGA.LN.A4A1.Tumor.RNAseq.ffd
589419 198953
TCGA.LN.A4A3.Tumor.RNAseq.7ab TCGA.LN.A4A4.Tumor.RNAseq.fc5
330219 345745
TCGA.LN.A4A5.Tumor.RNAseq.11e TCGA.LN.A4A8.Tumor.RNAseq.e2a
167082 169882
TCGA.LN.A4A9.Tumor.RNAseq.cc6 TCGA.LN.A4MQ.Tumor.RNAseq.5b4
498383 210714
TCGA.LN.A5U5.Tumor.RNAseq.ef4 TCGA.LN.A5U6.Tumor.RNAseq.c00
208664 205554
TCGA.LN.A5U7.Tumor.RNAseq.88b TCGA.LN.A7HV.Tumor.RNAseq.504
111690 0
TCGA.LN.A7HW.Tumor.RNAseq.1ad TCGA.LN.A7HX.Tumor.RNAseq.99e
0 0
TCGA.LN.A7HY.Tumor.RNAseq.f7b TCGA.LN.A7HZ.Tumor.RNAseq.740
0 0
TCGA.LN.A8HZ.Tumor.RNAseq.628 TCGA.LN.A8I0.Tumor.RNAseq.b5f
0 0
TCGA.LN.A8I1.Tumor.RNAseq.838 TCGA.LN.A9FO.Tumor.RNAseq.ae5
0 0
TCGA.LN.A9FP.Tumor.RNAseq.350 TCGA.LN.A9FQ.Tumor.RNAseq.082
0 0
TCGA.LN.A9FR.Tumor.RNAseq.304 TCGA.M9.A5M8.Tumor.RNAseq.4ef
0 233106
TCGA.Q9.A6FW.Tumor.RNAseq.d88 TCGA.R6.A6DN.Tumor.RNAseq.7ed
472304 197164
TCGA.R6.A6DQ.Tumor.RNAseq.a41 TCGA.R6.A6KZ.Tumor.RNAseq.22d
302461 188388
TCGA.R6.A6L4.Tumor.RNAseq.19e TCGA.R6.A6XG.Tumor.RNAseq.fde
242143 0
TCGA.R6.A6XQ.Tumor.RNAseq.ce6 TCGA.R6.A6Y0.Tumor.RNAseq.815
0 0
TCGA.R6.A8W5.Tumor.RNAseq.300 TCGA.R6.A8W8.Tumor.RNAseq.fb2
0 2269286
TCGA.R6.A8WC.Tumor.RNAseq.64b TCGA.R6.A8WG.Tumor.RNAseq.cb5
2118504 0
TCGA.RE.A7BO.Tumor.RNAseq.e67 TCGA.S8.A6BV.Tumor.RNAseq.86b
0 215598
TCGA.S8.A6BW.Tumor.RNAseq.802 TCGA.V5.A7RB.Tumor.RNAseq.2c7
136204 0
TCGA.V5.A7RC.Tumor.RNAseq.15e TCGA.V5.A7RC.Tumor.RNAseq.5f5
0 0
TCGA.V5.A7RE.Normal.RNAseq.848 TCGA.V5.A7RE.Tumor.RNAseq.7c1
0 0
TCGA.V5.AASV.Tumor.RNAseq.2bb TCGA.V5.AASW.Tumor.RNAseq.a2a
0 0
TCGA.V5.AASX.Normal.RNAseq.e7e TCGA.V5.AASX.Tumor.RNAseq.0c4
0 0
TCGA.VR.A8EO.Tumor.RNAseq.e76 TCGA.VR.A8EP.Tumor.RNAseq.afe
0 0
TCGA.VR.A8EQ.Tumor.RNAseq.350 TCGA.VR.A8ER.Tumor.RNAseq.a53
0 0
TCGA.VR.A8ET.Tumor.RNAseq.d5b TCGA.VR.A8EU.Tumor.RNAseq.421
0 0
TCGA.VR.A8EW.Tumor.RNAseq.a6b TCGA.VR.A8EX.Tumor.RNAseq.547
0 0
TCGA.VR.A8EY.Tumor.RNAseq.48e TCGA.VR.A8EZ.Tumor.RNAseq.48d
0 0
TCGA.VR.A8Q7.Tumor.RNAseq.053 TCGA.VR.AA4D.Tumor.RNAseq.325
0 0
TCGA.VR.AA4G.Tumor.RNAseq.52c TCGA.VR.AA7I.Tumor.RNAseq.1a6
0 0
TCGA.XP.A8T6.Tumor.RNAseq.5ca TCGA.XP.A8T8.Tumor.RNAseq.2d9
0 0
TCGA.Z6.A8JD.Tumor.RNAseq.d53 TCGA.Z6.A8JE.Tumor.RNAseq.e1a
0 0
TCGA.Z6.A9VB.Tumor.RNAseq.f2b TCGA.Z6.AAPN.Tumor.RNAseq.64f
0 0
TCGA.ZR.A9CJ.Tumor.RNAseq.d6f
0 1. Removal of Phylum NA features
# show ranks
phyloseq::rank_names(phylo.data.tcga)[1] "Kingdom" "Phylum" "Class" "Order" "Family" "Genus" # table of features for each phylum
table(phyloseq::tax_table(phylo.data.tcga)[,"Phylum"], exclude=NULL)
Acidobacteria Actinobacteria
5 142
Aquificae Bacteroidetes
1 62
candidate division NC10 Candidatus Saccharibacteria
1 3
Chloroflexi Cyanobacteria
1 11
Deinococcus-Thermus Euryarchaeota
12 1
Firmicutes Fusobacteria
181 5
Gemmatimonadetes Nitrospirae
1 1
Planctomycetes Proteobacteria
6 321
Spirochaetes Tenericutes
6 14
Verrucomicrobia
5 Note that no taxa were labels as NA so none were removed.
2. Computation of total and average prevalence in each Phylum
# compute prevalence of each feature
prevdf <- apply(X=phyloseq::otu_table(phylo.data.tcga),
MARGIN= ifelse(phyloseq::taxa_are_rows(phylo.data.tcga), yes=1, no=2),
FUN=function(x){sum(x>0)})
# store as data.frame with labels
prevdf <- data.frame(Prevalence=prevdf,
TotalAbundance=phyloseq::taxa_sums(phylo.data.tcga),
phyloseq::tax_table(phylo.data.tcga))Compute the totals and averages abundances.
totals <- plyr::ddply(prevdf, "Phylum",
function(df1){
A <- cbind(mean(df1$Prevalence), sum(df1$Prevalence))
colnames(A) <- c("Average", "Total")
A
}
) # end
totals Phylum Average Total
1 Acidobacteria 6.600000 33
2 Actinobacteria 32.415493 4603
3 Aquificae 3.000000 3
4 Bacteroidetes 20.500000 1271
5 candidate division NC10 4.000000 4
6 Candidatus Saccharibacteria 27.666667 83
7 Chloroflexi 5.000000 5
8 Cyanobacteria 6.545455 72
9 Deinococcus-Thermus 23.583333 283
10 Euryarchaeota 6.000000 6
11 Firmicutes 21.364641 3867
12 Fusobacteria 26.000000 130
13 Gemmatimonadetes 13.000000 13
14 Nitrospirae 6.000000 6
15 Planctomycetes 10.833333 65
16 Proteobacteria 32.688474 10493
17 Spirochaetes 5.333333 32
18 Tenericutes 3.214286 45
19 Verrucomicrobia 12.600000 63Any of the taxa under a total of 100 may be suspect. First, we will remove the taxa that are clearly too low in abundance (<=3).
filterPhyla <- totals$Phylum[totals$Total <= 3, drop=T] # drop allows some of the attributes to be removed
phylo.data1 <- phyloseq::subset_taxa(phylo.data.tcga, !Phylum %in% filterPhyla)
phylo.data1phyloseq-class experiment-level object
otu_table() OTU Table: [ 778 taxa and 173 samples ]
sample_data() Sample Data: [ 173 samples by 43 sample variables ]
tax_table() Taxonomy Table: [ 778 taxa by 6 taxonomic ranks ]Next, we explore the taxa in more detail next as we move to remove some of these low abundance taxa.
3. Removal Phyla with 0.1% or less of all samples (prevalence filtering)
prevdf1 <- subset(prevdf, Phylum %in% phyloseq::get_taxa_unique(phylo.data1, "Phylum"))4. Total count computation
# already done above ()5. Threshold identification
ggplot(prevdf1, aes(TotalAbundance+1,
Prevalence/nsamples(phylo.data.tcga))) +
geom_hline(yintercept=0.01, alpha=0.5, linetype=2)+
geom_point(size=2, alpha=0.75) +
scale_x_log10()+
labs(x="Total Abundance", y="Prevalance [Frac. Samples]")+
facet_wrap(.~Phylum) + theme(legend.position = "none")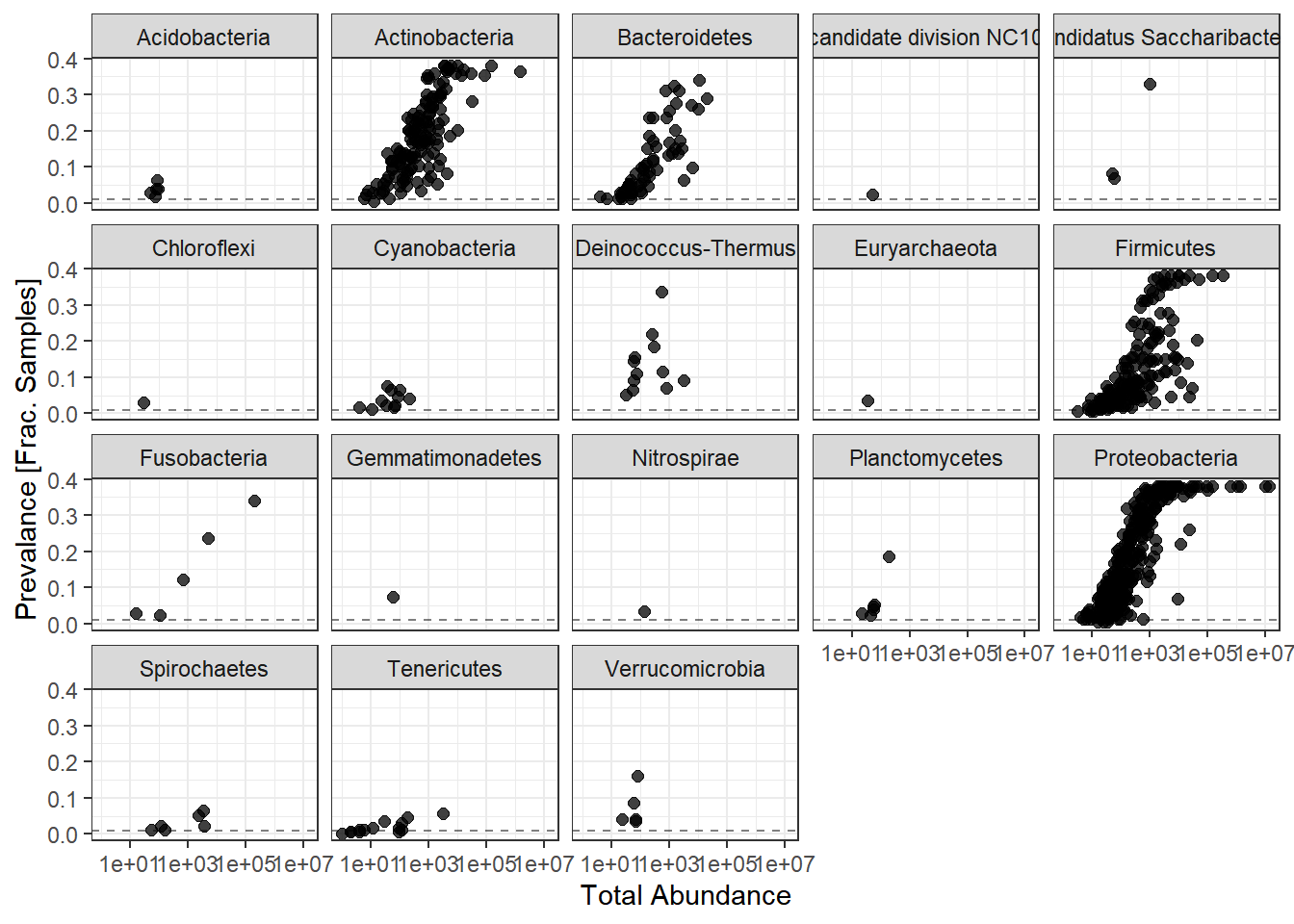
Note: for plotting purposes, a \(+1\) was added to all TotalAbundances to avoid a taking the log of 0.
Next, we define a prevalence threshold, that way the taxa can be pruned to a prespecified level. In this study, we used 0.0001 (0.01%) of total samples.
prevalenceThreshold <- 0.0001*(phyloseq::nsamples(phylo.data.tcga))
prevalenceThreshold[1] 0.0173# execute the filtering to this level
keepTaxa <- rownames(prevdf1)[(prevdf1$Prevalence >= prevalenceThreshold)]
phylo.data2 <- phyloseq::prune_taxa(keepTaxa, phylo.data1)6. Merge taxa (to genus level)
genusNames <- phyloseq::get_taxa_unique(phylo.data2, "Genus")
#phylo.data3 <- merge_taxa(phylo.data2, genusNames, genusNames[which.max(taxa_sums(phylo.data2)[genusNames])])
# How many genera would be present after filtering?
length(phyloseq::get_taxa_unique(phylo.data2, taxonomic.rank = "Genus"))[1] 380phylo.data3 = phyloseq::tax_glom(phylo.data2, "Genus", NArm = TRUE)7. Removal of Genera Contaminants
a <- taxa_names(phylo.data3)
conTaxa <- c("Pseudomonadales", "Comamonadaceae", "Rhizobiales", "Burkholderiales", "Paenibacillaceae", "Staphylococcus epidermidis", "Propionibacterium acnes", "Escherichia", "Bacillaceae")
i <- 1
K <- 0
for(i in 1:length(conTaxa)){
kT <- a[a %like% conTaxa[i]]
K <- c(K, kT)
}
b <- !a %in% K
phylo.data3 <- phyloseq::prune_taxa(b, phylo.data3)8. Relative Adbundance Plot
plot_abundance = function(physeq, title = "", ylab="Abundance"){
mphyseq = phyloseq::psmelt(physeq)
mphyseq <- subset(mphyseq, Abundance > 0)
ggplot(data = mphyseq, aes(x=SampleType_Level2, y=Abundance)) +
geom_violin(fill = NA) +
geom_point(size = 1, alpha = 0.9,
position = position_jitter(width = 0.3)) +
scale_y_log10()+
labs(y=ylab)+
theme(legend.position="none")
}
# Transform to relative abundance. Save as new object.
phylo.data3ra = transform_sample_counts(phylo.data3, function(x){x / sum(x)})
plotBefore = plot_abundance(phylo.data3, ylab="Abundance prior to transformation")
plotAfter = plot_abundance(phylo.data3ra, ylab="Relative Abundance")
# Combine each plot into one graphic.
plotBefore + plotAfter + plot_layout(nrow=2)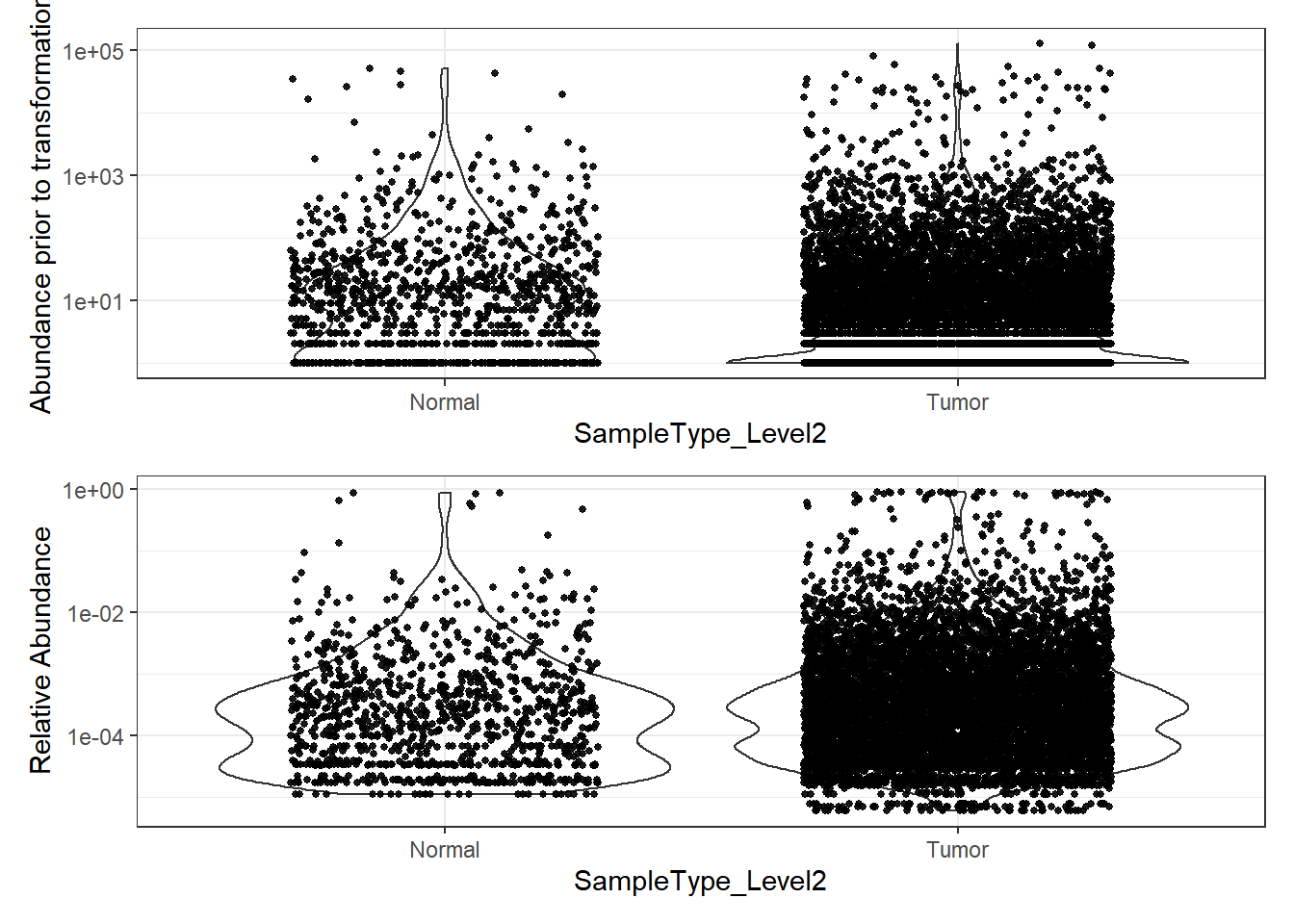
9. Plotting Abundance
Abundance by Phylum
plot_abundance = function(physeq, title = "", Facet = "Phylum",
ylab="Abundance"){
mphyseq = phyloseq::psmelt(physeq)
mphyseq <- subset(mphyseq, Abundance > 0)
ggplot(data = mphyseq, aes(x=SampleType_Level2, y=Abundance)) +
geom_violin(fill = NA) +
geom_point(size = 1, alpha = 0.9,
position = position_jitter(width = 0.3)) +
facet_wrap(facets = Facet) + scale_y_log10()+
labs(y=ylab)+
theme(legend.position="none")
}
plotBefore = plot_abundance(phylo.data3, ylab="Abundance prior to transformation")
plotAfter = plot_abundance(phylo.data3ra, ylab="Relative Abundance")
# Combine each plot into one graphic.
plotBefore + plotAfter + plot_layout(nrow=2)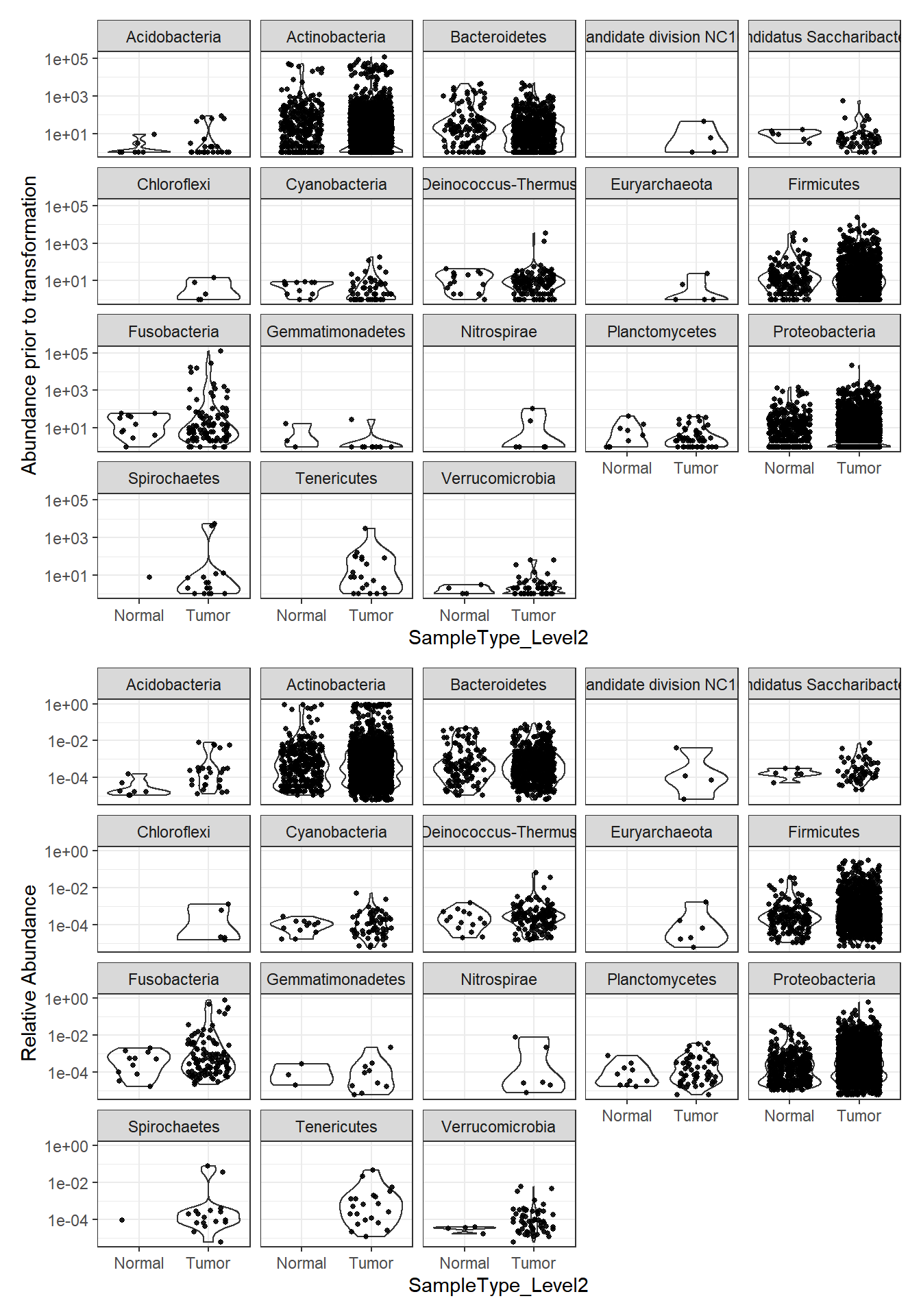
Phylum: Fusobacteria
plot_abundance = function(physeq, title = "", Facet = "Genus", ylab="Abundance"){
mphyseq = phyloseq::subset_taxa(physeq, Phylum %in% "Fusobacteria")
mphyseq <- phyloseq::psmelt(mphyseq)
mphyseq <- subset(mphyseq, Abundance > 0)
ggplot(data = mphyseq, aes(x=SampleType_Level2, y=Abundance)) +
geom_violin(fill = NA) +
geom_point(size = 1, alpha = 0.9,
position = position_jitter(width = 0.3)) +
facet_wrap(facets = Facet) + scale_y_log10()+
labs(y=ylab)+
theme(legend.position="none")
}
plotBefore = plot_abundance(phylo.data3,
ylab="Abundance prior to transformation")
plotAfter = plot_abundance(phylo.data3ra,
ylab="Relative Abundance")
plotBefore + plotAfter + plot_layout(nrow=2)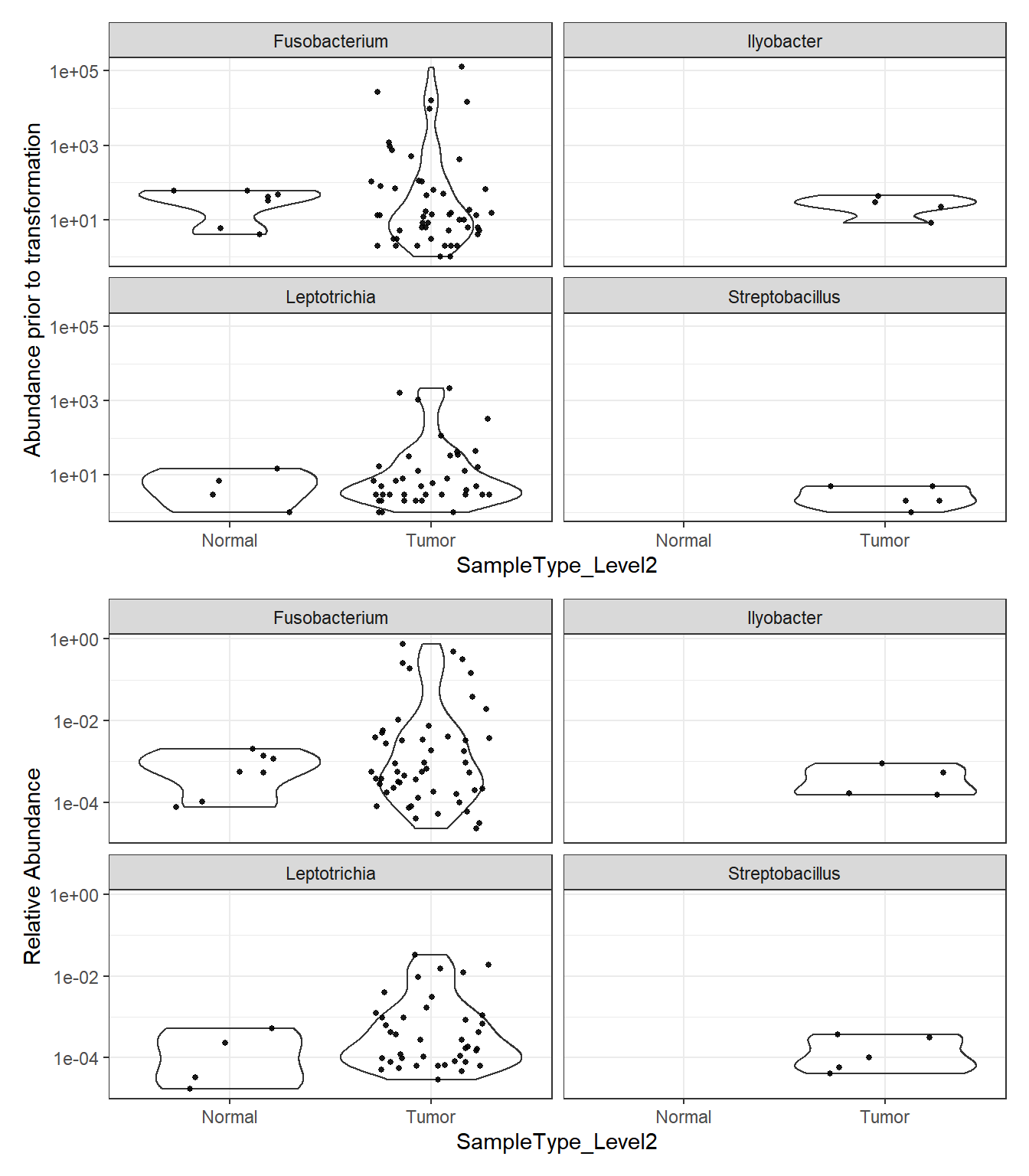
WGS Data: Taxonomic Filtering
0. Sample Reads, Totals, and Rarifying
sampleReads <- sample_sums(phylo.data.tcga.WGS)
# Total quality Reads
sum(sampleReads)[1] 3817495# Average reads
mean(sampleReads)[1] 27463.99# max sequencing depth
max(sampleReads)[1] 559963# rarified to an even depth of
phylo.data.tcga <- phylo.data.tcga.WGS #rarefy_even_depth(phylo.data.tcga.WGS, replace = T, rngseed = 20200923)
# even depth of:
sample_sums(phylo.data.tcga)TCGA.IG.A3I8.Normal.WGS.222 TCGA.IG.A3I8.Normal.WGS.a07
412 447
TCGA.IG.A3I8.Tumor.WGS.d45 TCGA.IG.A3QL.Normal.WGS.e64
1576 42617
TCGA.IG.A3QL.Tumor.WGS.da1 TCGA.IG.A3Y9.Normal.WGS.dbf
6268 34517
TCGA.IG.A3Y9.Tumor.WGS.c2e TCGA.IG.A3YA.Normal.WGS.30a
24967 40161
TCGA.IG.A3YA.Tumor.WGS.d20 TCGA.IG.A3YB.Normal.WGS.be8
23320 26565
TCGA.IG.A3YB.Tumor.WGS.6b5 TCGA.IG.A3YB.Tumor.WGS.aac
22708 52711
TCGA.IG.A3YC.Normal.WGS.42f TCGA.IG.A3YC.Tumor.WGS.a12
23137 15430
TCGA.IG.A4P3.Normal.WGS.905 TCGA.IG.A4P3.Tumor.WGS.a5b
2740 2206
TCGA.IG.A4QT.Normal.WGS.555 TCGA.IG.A4QT.Tumor.WGS.4bf
4023 4020
TCGA.IG.A50L.Normal.WGS.076 TCGA.IG.A50L.Tumor.WGS.3e7
5095 289811
TCGA.IG.A51D.Normal.WGS.780 TCGA.IG.A51D.Tumor.WGS.c42
12504 12472
TCGA.IG.A5B8.Normal.WGS.bbd TCGA.IG.A5B8.Tumor.WGS.948
6999 7698
TCGA.IG.A5S3.Normal.WGS.035 TCGA.IG.A5S3.Tumor.WGS.736
214 290
TCGA.IG.A97I.Normal.WGS.900 TCGA.IG.A97I.Tumor.WGS.9b8
33031 0
TCGA.JY.A93C.Tumor.WGS.f40 TCGA.L5.A43C.Normal.WGS.311
35048 21546
TCGA.L5.A43C.Tumor.WGS.f5b TCGA.L5.A43E.Normal.WGS.280
12862 40554
TCGA.L5.A43E.Normal.WGS.56f TCGA.L5.A43E.Tumor.WGS.70d
47251 11531
TCGA.L5.A43H.Normal.WGS.d67 TCGA.L5.A43H.Tumor.WGS.f8c
49147 28611
TCGA.L5.A43I.Normal.WGS.293 TCGA.L5.A43I.Tumor.WGS.dee
40707 8126
TCGA.L5.A43J.Normal.WGS.e99 TCGA.L5.A43J.Tumor.WGS.ed6
26652 6627
TCGA.L5.A43M.Normal.WGS.ec5 TCGA.L5.A43M.Tumor.WGS.4e3
369 1478
TCGA.L5.A4OE.Normal.WGS.a69 TCGA.L5.A4OE.Tumor.WGS.498
12714 2948
TCGA.L5.A4OF.Normal.WGS.798 TCGA.L5.A4OF.Normal.WGS.948
0 18579
TCGA.L5.A4OF.Tumor.WGS.0b3 TCGA.L5.A4OF.Tumor.WGS.3ee
4369 79921
TCGA.L5.A4OG.Normal.WGS.963 TCGA.L5.A4OG.Tumor.WGS.cef
165 260
TCGA.L5.A4OH.Normal.WGS.418 TCGA.L5.A4OH.Tumor.WGS.ba8
2963 650
TCGA.L5.A4OI.Normal.WGS.600 TCGA.L5.A4OI.Tumor.WGS.61f
42067 5504
TCGA.L5.A4OJ.Normal.WGS.3de TCGA.L5.A4OJ.Normal.WGS.9dc
86175 63
TCGA.L5.A4OJ.Tumor.WGS.b81 TCGA.L5.A4OM.Normal.WGS.6cb
114841 220
TCGA.L5.A4OM.Tumor.WGS.206 TCGA.L5.A4ON.Normal.WGS.076
184 1378
TCGA.L5.A4ON.Tumor.WGS.9bb TCGA.L5.A4OP.Normal.WGS.6e4
316 169
TCGA.L5.A4OP.Tumor.WGS.940 TCGA.L5.A4OR.Normal.WGS.440
96 0
TCGA.L5.A4OR.Tumor.WGS.8a6 TCGA.L5.A4OS.Normal.WGS.643
0 14416
TCGA.L5.A4OS.Tumor.WGS.5c0 TCGA.L5.A4OT.Normal.WGS.2a1
112066 8373
TCGA.L5.A4OT.Tumor.WGS.7d4 TCGA.L5.A891.Normal.WGS.9fa
42262 269777
TCGA.L5.A891.Tumor.WGS.6a8 TCGA.L5.A8NE.Normal.WGS.da6
559963 25856
TCGA.L5.A8NE.Tumor.WGS.d6f TCGA.L5.A8NN.Normal.WGS.5ef
0 0
TCGA.L5.A8NN.Tumor.WGS.d94 TCGA.L7.A56G.Normal.WGS.706
0 2946
TCGA.L7.A56G.Tumor.WGS.8e8 TCGA.L7.A6VZ.Normal.WGS.58c
173707 0
TCGA.L7.A6VZ.Tumor.WGS.8fa TCGA.LN.A49K.Normal.WGS.46d
0 28461
TCGA.LN.A49K.Tumor.WGS.acf TCGA.LN.A49L.Normal.WGS.99b
15830 1793
TCGA.LN.A49L.Normal.WGS.c79 TCGA.LN.A49L.Tumor.WGS.a95
37639 30608
TCGA.LN.A49L.Tumor.WGS.a9a TCGA.LN.A49M.Normal.WGS.86f
8660 6224
TCGA.LN.A49M.Normal.WGS.d6f TCGA.LN.A49M.Tumor.WGS.821
0 5518
TCGA.LN.A49M.Tumor.WGS.dae TCGA.LN.A49N.Normal.WGS.aa8
0 69332
TCGA.LN.A49N.Tumor.WGS.cc2 TCGA.LN.A49O.Normal.WGS.0e9
39430 78468
TCGA.LN.A49O.Tumor.WGS.e46 TCGA.LN.A49P.Normal.WGS.09a
33726 57063
TCGA.LN.A49P.Tumor.WGS.bc9 TCGA.LN.A49R.Normal.WGS.427
21648 898
TCGA.LN.A49R.Tumor.WGS.94a TCGA.LN.A49S.Normal.WGS.a7e
14897 12081
TCGA.LN.A49S.Tumor.WGS.f17 TCGA.LN.A49U.Normal.WGS.bc2
8885 5318
TCGA.LN.A49U.Tumor.WGS.c07 TCGA.LN.A49V.Normal.WGS.693
51386 22949
TCGA.LN.A49V.Tumor.WGS.331 TCGA.LN.A49W.Normal.WGS.cfd
34529 3514
TCGA.LN.A49W.Tumor.WGS.30b TCGA.LN.A49X.Normal.WGS.36d
2374 5414
TCGA.LN.A49X.Tumor.WGS.e3d TCGA.LN.A49Y.Normal.WGS.859
3022 7802
TCGA.LN.A49Y.Normal.WGS.902 TCGA.LN.A49Y.Tumor.WGS.61d
2661 526
TCGA.LN.A4A1.Normal.WGS.774 TCGA.LN.A4A1.Tumor.WGS.de6
13062 0
TCGA.LN.A4A2.Normal.WGS.851 TCGA.LN.A4A2.Tumor.WGS.2f7
16038 14355
TCGA.LN.A4A3.Normal.WGS.bee TCGA.LN.A4A3.Tumor.WGS.e04
3899 172865
TCGA.LN.A4A4.Normal.WGS.200 TCGA.LN.A4A4.Normal.WGS.c72
6580 33938
TCGA.LN.A4A4.Tumor.WGS.330 TCGA.LN.A4A4.Tumor.WGS.8f2
0 1302
TCGA.LN.A4A6.Normal.WGS.298 TCGA.LN.A4A6.Tumor.WGS.113
2142 1280
TCGA.LN.A4A8.Normal.WGS.3df TCGA.LN.A4A8.Tumor.WGS.fd3
3468 1688
TCGA.LN.A4MQ.Normal.WGS.51c TCGA.LN.A4MQ.Tumor.WGS.beb
9082 3647
TCGA.LN.A4MR.Normal.WGS.a1b TCGA.LN.A4MR.Tumor.WGS.6c3
1810 2753
TCGA.LN.A5U5.Normal.WGS.25f TCGA.LN.A5U5.Normal.WGS.e87
3029 21262
TCGA.LN.A5U5.Tumor.WGS.d38 TCGA.LN.A5U5.Tumor.WGS.ee1
2427 181294
TCGA.LN.A8I1.Normal.WGS.e4b TCGA.LN.A8I1.Tumor.WGS.70b
16143 23999
TCGA.LN.A9FQ.Normal.WGS.432 TCGA.LN.A9FQ.Tumor.WGS.a06
25536 0
TCGA.V5.A7RC.Normal.WGS.a0c TCGA.V5.A7RC.Tumor.WGS.050
35844 0
TCGA.V5.A7RC.Tumor.WGS.3ce
0 1. Removal of Phylum NA features
# show ranks
rank_names(phylo.data.tcga)[1] "Kingdom" "Phylum" "Class" "Order" "Family" "Genus" # table of features for each phylum
table(tax_table(phylo.data.tcga)[,"Phylum"], exclude=NULL)
Acidobacteria Actinobacteria
5 142
Aquificae Bacteroidetes
1 62
candidate division NC10 Candidatus Saccharibacteria
1 3
Chloroflexi Cyanobacteria
1 11
Deinococcus-Thermus Euryarchaeota
12 1
Firmicutes Fusobacteria
181 5
Gemmatimonadetes Nitrospirae
1 1
Planctomycetes Proteobacteria
6 321
Spirochaetes Tenericutes
6 14
Verrucomicrobia
5 Note that no taxa were labels as NA so none were removed.
2. Computation of total and average prevalence in each Phylum
# compute prevalence of each feature
prevdf <- apply(X=otu_table(phylo.data.tcga),
MARGIN= ifelse(taxa_are_rows(phylo.data.tcga), yes=1, no=2),
FUN=function(x){sum(x>0)})
# store as data.frame with labels
prevdf <- data.frame(Prevalence=prevdf,
TotalAbundance=taxa_sums(phylo.data.tcga),
tax_table(phylo.data.tcga))Compute the totals and averages abundances.
totals <- plyr::ddply(prevdf, "Phylum",
function(df1){
A <- cbind(mean(df1$Prevalence), sum(df1$Prevalence))
colnames(A) <- c("Average", "Total")
A
}
) # end
totals Phylum Average Total
1 Acidobacteria 2.400000 12
2 Actinobacteria 13.190141 1873
3 Aquificae 2.000000 2
4 Bacteroidetes 14.790323 917
5 candidate division NC10 3.000000 3
6 Candidatus Saccharibacteria 61.666667 185
7 Chloroflexi 2.000000 2
8 Cyanobacteria 13.454545 148
9 Deinococcus-Thermus 4.833333 58
10 Euryarchaeota 1.000000 1
11 Firmicutes 13.696133 2479
12 Fusobacteria 25.600000 128
13 Gemmatimonadetes 6.000000 6
14 Nitrospirae 4.000000 4
15 Planctomycetes 8.500000 51
16 Proteobacteria 13.875389 4454
17 Spirochaetes 11.166667 67
18 Tenericutes 5.500000 77
19 Verrucomicrobia 5.400000 27Any of the taxa under a total of 100 may be suspect. First, we will remove the taxa that are clearly too low in abundance (<=3).
filterPhyla <- totals$Phylum[totals$Total <= 3, drop=T] # drop allows some of the attributes to be removed
phylo.data1 <- subset_taxa(phylo.data.tcga, !Phylum %in% filterPhyla)
phylo.data1phyloseq-class experiment-level object
otu_table() OTU Table: [ 775 taxa and 139 samples ]
sample_data() Sample Data: [ 139 samples by 43 sample variables ]
tax_table() Taxonomy Table: [ 775 taxa by 6 taxonomic ranks ]Next, we explore the taxa in more detail next as we move to remove some of these low abundance taxa.
3. Removal Phyla with 0.1% or less of all samples (prevalence filtering)
prevdf1 <- subset(prevdf, Phylum %in% get_taxa_unique(phylo.data1, "Phylum"))4. Total count computation
# already done above ()5. Threshold identification
ggplot(prevdf1, aes(TotalAbundance+1,
Prevalence/nsamples(phylo.data.tcga))) +
geom_hline(yintercept=0.01, alpha=0.5, linetype=2)+
geom_point(size=2, alpha=0.75) +
scale_x_log10()+
labs(x="Total Abundance", y="Prevalance [Frac. Samples]")+
facet_wrap(.~Phylum) + theme(legend.position = "none")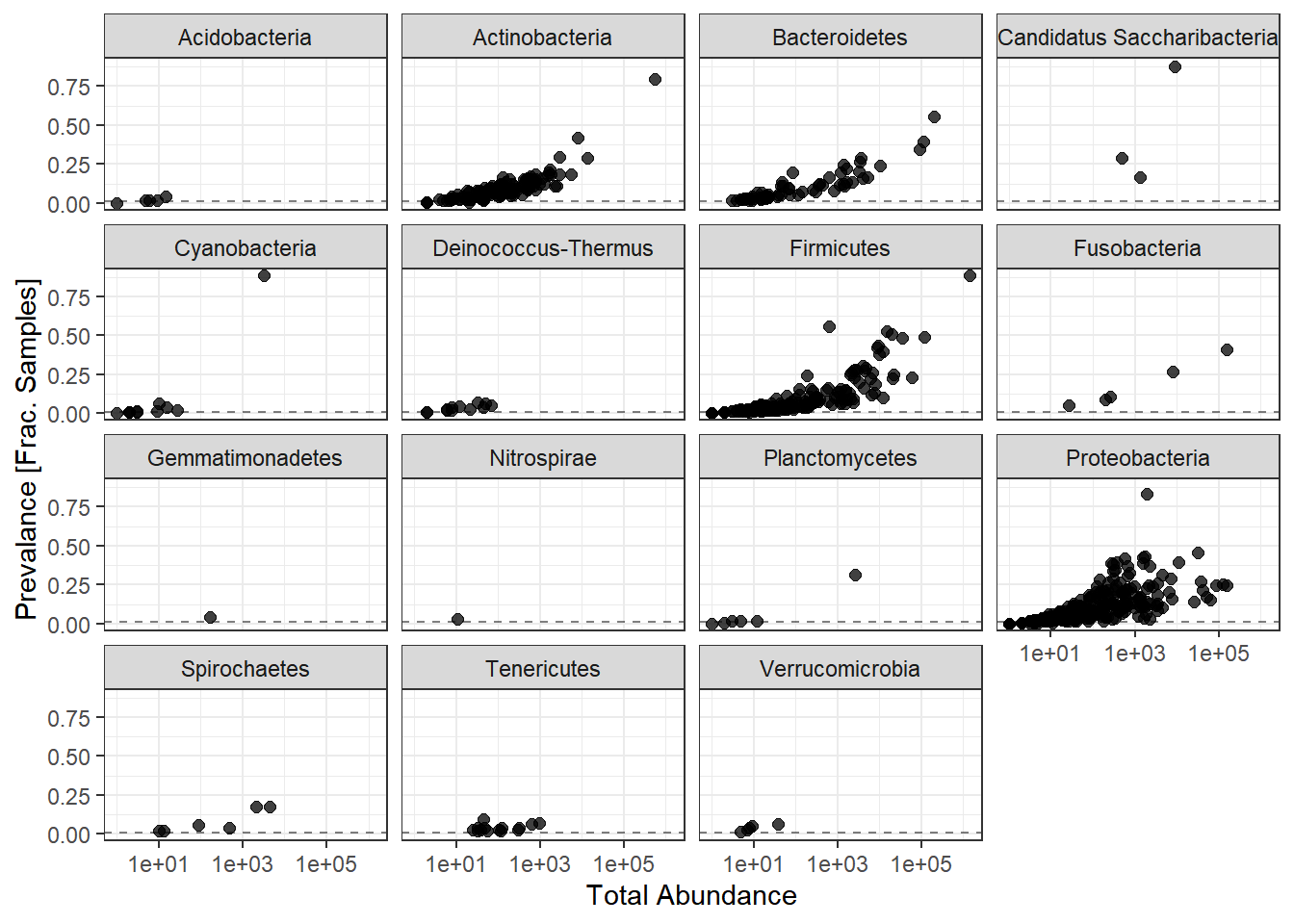
Note: for plotting purposes, a \(+1\) was added to all TotalAbundances to avoid a taking the log of 0.
Next, we define a prevalence threshold, that way the taxa can be pruned to a prespecified level. In this study, we used 0.0001 (0.01%) of total samples.
prevalenceThreshold <- 0.0001*nsamples(phylo.data.tcga)
prevalenceThreshold[1] 0.0139# execute the filtering to this level
keepTaxa <- rownames(prevdf1)[(prevdf1$Prevalence >= prevalenceThreshold)]
phylo.data2 <- prune_taxa(keepTaxa, phylo.data1)6. Merge taxa (to genus level)
genusNames <- get_taxa_unique(phylo.data2, "Genus")
#phylo.data3 <- merge_taxa(phylo.data2, genusNames, genusNames[which.max(taxa_sums(phylo.data2)[genusNames])])
# How many genera would be present after filtering?
length(get_taxa_unique(phylo.data2, taxonomic.rank = "Genus"))[1] 373phylo.data3 = tax_glom(phylo.data2, "Genus", NArm = TRUE)7. Removal of Genera Contaminants
a <- taxa_names(phylo.data3)
conTaxa <- c("Pseudomonadales", "Comamonadaceae", "Rhizobiales", "Burkholderiales", "Paenibacillaceae", "Staphylococcus epidermidis", "Propionibacterium acnes", "Escherichia", "Bacillaceae")
i <- 1
K <- 0
for(i in 1:length(conTaxa)){
kT <- a[a %like% conTaxa[i]]
K <- c(K, kT)
}
b <- !a %in% K
phylo.data3 <- phyloseq::prune_taxa(b, phylo.data3)8. Relative Adbundance Plot
plot_abundance = function(physeq, title = "", ylab="Abundance"){
mphyseq = psmelt(physeq)
mphyseq <- subset(mphyseq, Abundance > 0)
ggplot(data = mphyseq, aes(x=SampleType_Level2, y=Abundance)) +
geom_violin(fill = NA) +
geom_point(size = 1, alpha = 0.9,
position = position_jitter(width = 0.3)) +
scale_y_log10()+
labs(y=ylab)+
theme(legend.position="none")
}
# Transform to relative abundance. Save as new object.
phylo.data3ra = transform_sample_counts(phylo.data3, function(x){x / sum(x)})
plotBefore = plot_abundance(phylo.data3, ylab="Abundance prior to transformation")
plotAfter = plot_abundance(phylo.data3ra, ylab="Relative Abundance")
# Combine each plot into one graphic.
plotBefore + plotAfter + plot_layout(nrow=2)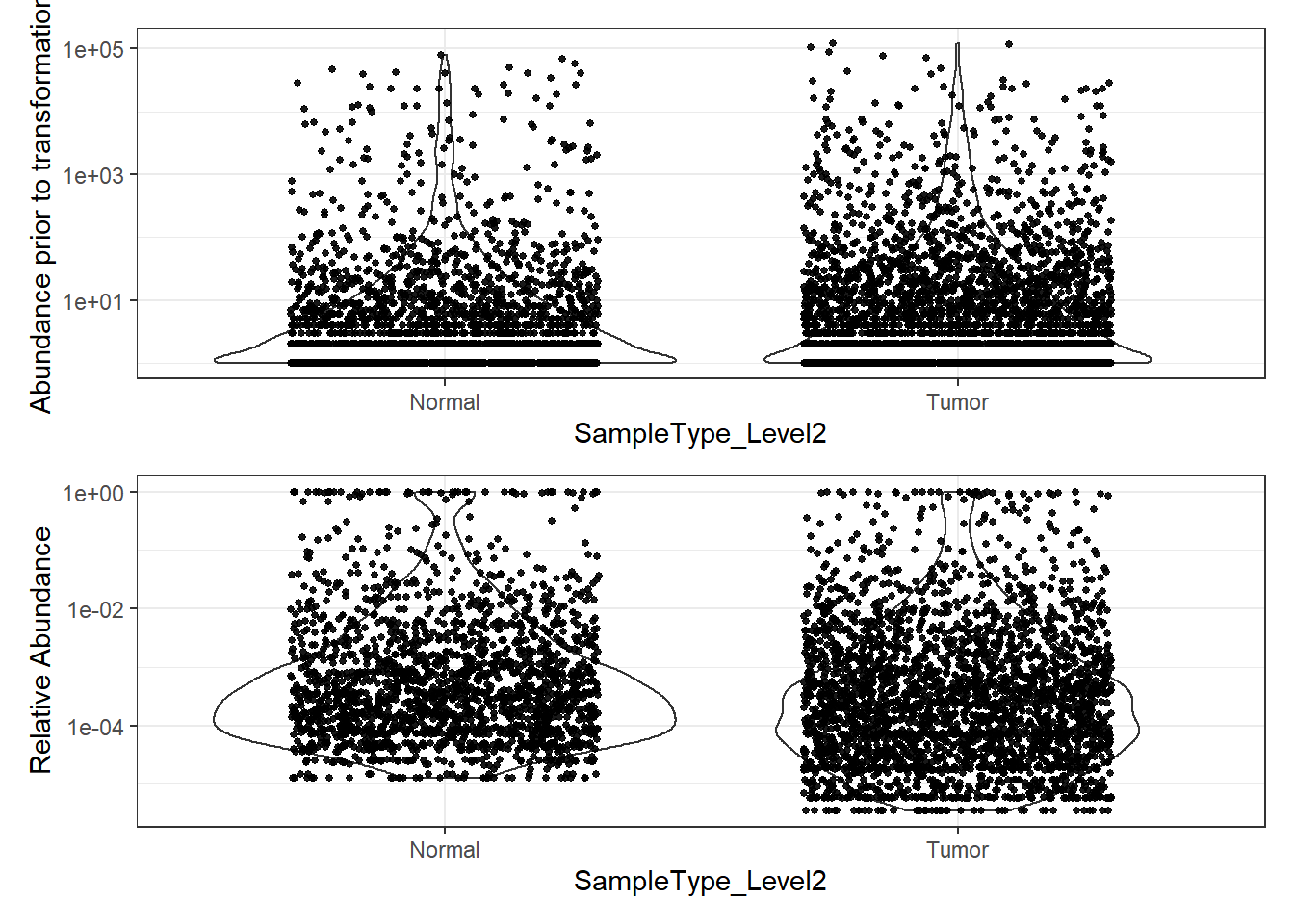
9. Plotting Abundance
Abundance by Phylum
plot_abundance = function(physeq, title = "", Facet = "Phylum", ylab="Abundance"){
mphyseq <- phyloseq::psmelt(physeq)
mphyseq <- subset(mphyseq, Abundance > 0)
ggplot(data = mphyseq, aes(x=SampleType_Level2, y=Abundance)) +
geom_violin(fill = NA) +
geom_point(size = 1, alpha = 0.9,
position = position_jitter(width = 0.3)) +
facet_wrap(facets = Facet) + scale_y_log10()+
labs(y=ylab)+
theme(legend.position="none")
}
plotBefore = plot_abundance(phylo.data3,
ylab="Abundance prior to transformation")
plotAfter = plot_abundance(phylo.data3ra,
ylab="Relative Abundance")
plotBefore + plotAfter + plot_layout(nrow=2)Warning in max(data$density): no non-missing arguments to max; returning -InfWarning: Computation failed in `stat_ydensity()`:
replacement has 1 row, data has 0Warning in max(data$density): no non-missing arguments to max; returning -InfWarning: Computation failed in `stat_ydensity()`:
replacement has 1 row, data has 0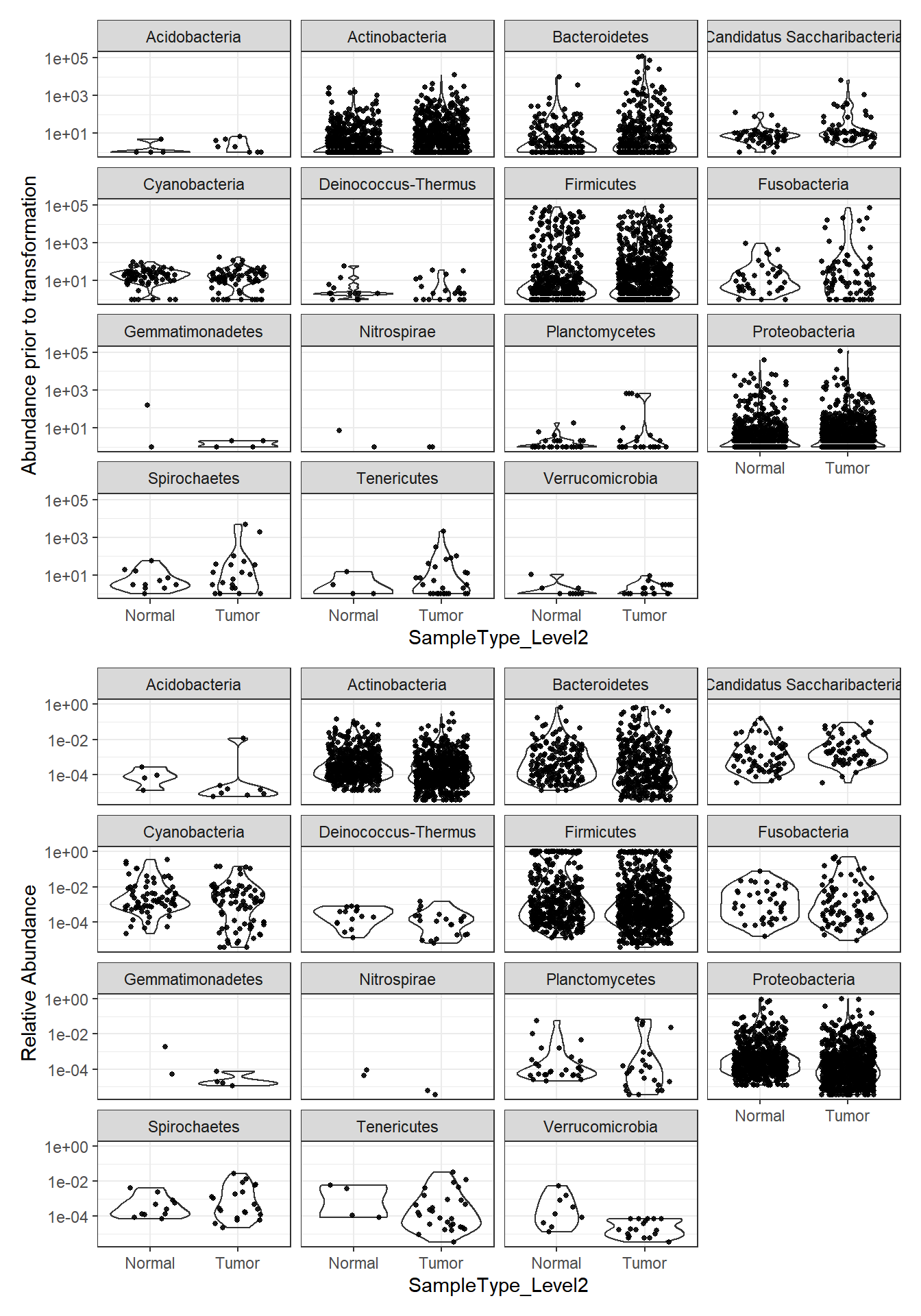
Phylum: Fusobacteria
plot_abundance = function(physeq, title = "", Facet = "Genus", ylab="Abundance"){
mphyseq = phyloseq::subset_taxa(physeq, Phylum %in% "Fusobacteria")
mphyseq <- phyloseq::psmelt(mphyseq)
mphyseq <- subset(mphyseq, Abundance > 0)
ggplot(data = mphyseq, aes(x=SampleType_Level2, y=Abundance)) +
geom_violin(fill = NA) +
geom_point(size = 1, alpha = 0.9,
position = position_jitter(width = 0.3)) +
facet_wrap(facets = Facet) + scale_y_log10()+
labs(y=ylab)+
theme(legend.position="none")
}
plotBefore = plot_abundance(phylo.data3,
ylab="Abundance prior to transformation")
plotAfter = plot_abundance(phylo.data3ra,
ylab="Relative Abundance")
plotBefore + plotAfter + plot_layout(nrow=2)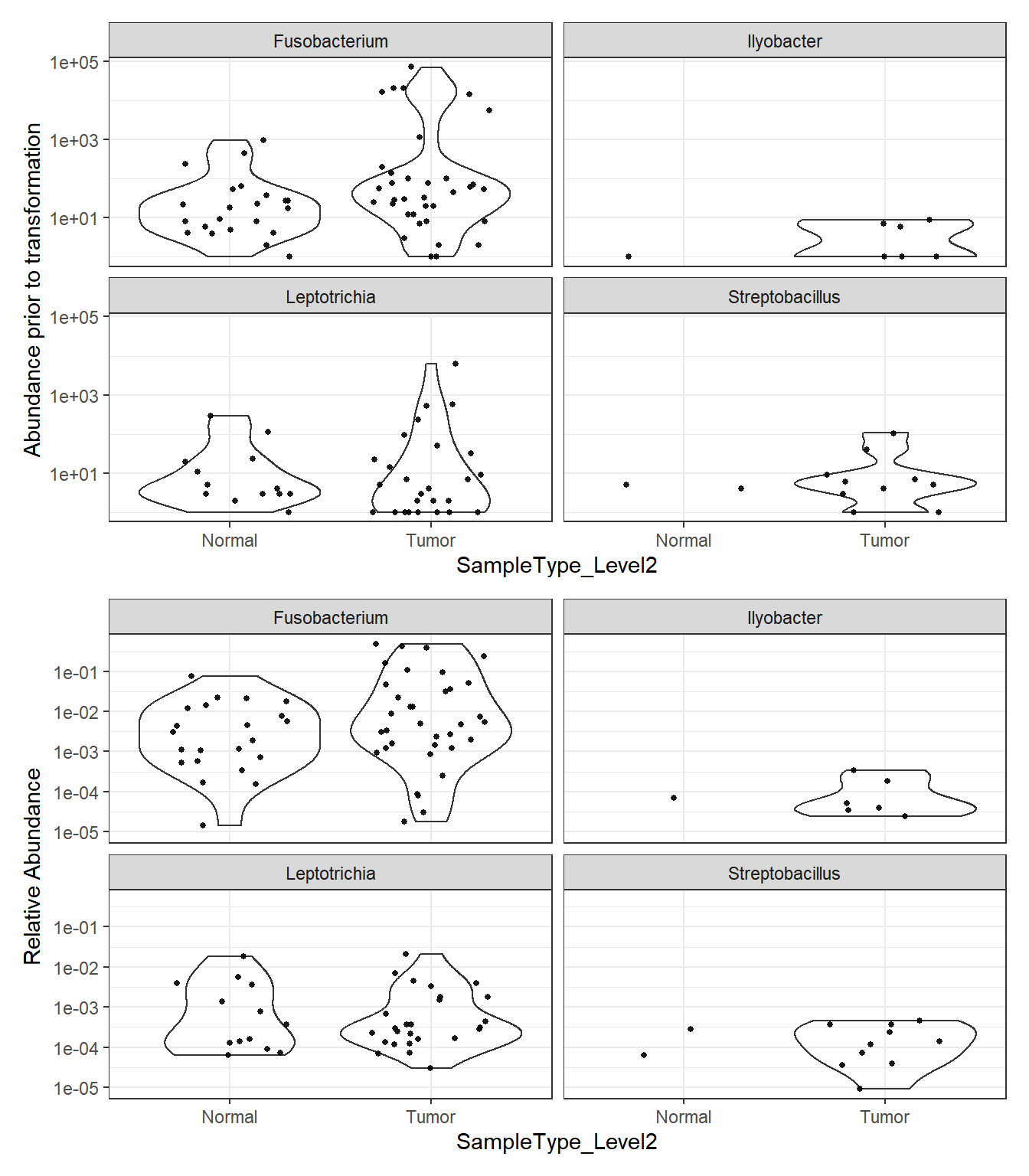
sessionInfo()R version 4.0.2 (2020-06-22)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 18363)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.1252
[2] LC_CTYPE=English_United States.1252
[3] LC_MONETARY=English_United States.1252
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.1252
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] car_3.0-8 carData_3.0-4 gvlma_1.0.0.3 patchwork_1.0.1
[5] viridis_0.5.1 viridisLite_0.3.0 gridExtra_2.3 xtable_1.8-4
[9] kableExtra_1.1.0 plyr_1.8.6 data.table_1.13.0 readxl_1.3.1
[13] forcats_0.5.0 stringr_1.4.0 dplyr_1.0.1 purrr_0.3.4
[17] readr_1.3.1 tidyr_1.1.1 tibble_3.0.3 ggplot2_3.3.2
[21] tidyverse_1.3.0 lmerTest_3.1-2 lme4_1.1-23 Matrix_1.2-18
[25] vegan_2.5-6 lattice_0.20-41 permute_0.9-5 phyloseq_1.32.0
[29] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] minqa_1.2.4 colorspace_1.4-1 rio_0.5.16
[4] ellipsis_0.3.1 rprojroot_1.3-2 XVector_0.28.0
[7] fs_1.5.0 rstudioapi_0.11 farver_2.0.3
[10] fansi_0.4.1 lubridate_1.7.9 xml2_1.3.2
[13] codetools_0.2-16 splines_4.0.2 knitr_1.29
[16] ade4_1.7-15 jsonlite_1.7.0 nloptr_1.2.2.2
[19] broom_0.7.0 cluster_2.1.0 dbplyr_1.4.4
[22] BiocManager_1.30.10 compiler_4.0.2 httr_1.4.2
[25] backports_1.1.7 assertthat_0.2.1 cli_2.0.2
[28] later_1.1.0.1 htmltools_0.5.0 tools_4.0.2
[31] igraph_1.2.5 gtable_0.3.0 glue_1.4.1
[34] reshape2_1.4.4 Rcpp_1.0.5 Biobase_2.48.0
[37] cellranger_1.1.0 vctrs_0.3.2 Biostrings_2.56.0
[40] multtest_2.44.0 ape_5.4 nlme_3.1-148
[43] iterators_1.0.12 xfun_0.19 openxlsx_4.1.5
[46] rvest_0.3.6 lifecycle_0.2.0 statmod_1.4.34
[49] zlibbioc_1.34.0 MASS_7.3-51.6 scales_1.1.1
[52] hms_0.5.3 promises_1.1.1 parallel_4.0.2
[55] biomformat_1.16.0 rhdf5_2.32.2 curl_4.3
[58] yaml_2.2.1 stringi_1.4.6 S4Vectors_0.26.1
[61] foreach_1.5.0 BiocGenerics_0.34.0 zip_2.0.4
[64] boot_1.3-25 rlang_0.4.7 pkgconfig_2.0.3
[67] evaluate_0.14 Rhdf5lib_1.10.1 labeling_0.3
[70] tidyselect_1.1.0 magrittr_1.5 R6_2.4.1
[73] IRanges_2.22.2 generics_0.0.2 DBI_1.1.0
[76] foreign_0.8-80 pillar_1.4.6 haven_2.3.1
[79] whisker_0.4 withr_2.2.0 mgcv_1.8-31
[82] abind_1.4-5 survival_3.2-3 modelr_0.1.8
[85] crayon_1.3.4 rmarkdown_2.5 grid_4.0.2
[88] blob_1.2.1 git2r_0.27.1 reprex_0.3.0
[91] digest_0.6.25 webshot_0.5.2 httpuv_1.5.4
[94] numDeriv_2016.8-1.1 stats4_4.0.2 munsell_0.5.0